Getting Started
Welcome
What is ScType?
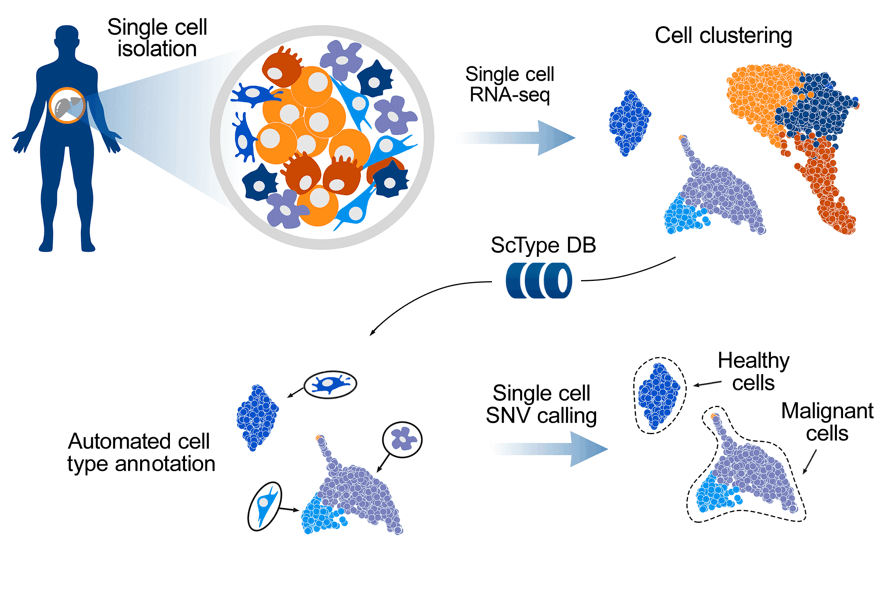
ScType is a tool for fully-automated cell type identification from single-cell RNA-seq data. ScType provides a complete pipeline for single-cell RNA-seq data analysis (including data processing, normalization and clustering) and cell-type annotation.
For more details, please refer to [to be filled]
- ScType requires only a scRNA-seq count matrix (either raw or normalized) as an input. The dimensionality of input data is unrestricted;
- ScType provides multiple methods for processing, analysis and visualization of scRNA-seq data;
- ScType implements an automated procedure for cell-type identification;
- ScType is a free tool with an open-source code available under GNU General Public License v3.0
ScType workflow
We highly recommend going through a step-by-step tour by clicking "How-to-use tour" button on the main page of the tool. This will guide you through the standard data processing workflow for scRNA-seq data analysis and cell type identification as well as introduce all the website functionality.
ScType workflow consists of the following steps:
- Input data quality control (as implemented in Seurat pipeline, latest version) to visually explore the data, including:
- Cell- based quality control (e.g. removing cells with unexpectedly high counts)
- Gene- based quality control (i.e. subsetting highly-variable genes)
- Data normalization and scaling (The default normalization is done using SCTransform, but other methods, such as log-normalization are available)
- Data clustering based on highly variable genes (default is Louvain clustering based on a shared nearest neighbor graph, but many other options are available)
- Differential expression analysis and cell marker identification.
- Automatic cell type identification and quantification
- Visualization of results (multiple options are available)
ScType input and output
ScType input
For easier user experience, three possible input file formats (Gene expression matrix, 10X genomics cellranger output, or SingleCellExperiment R object) are allowed in ScType. Therefore, the same input data can be provided in three alternative formats.
Note: The maximum input file size is 1GB.
a. Gene expression matrix
In the matrix format, gene-cell expression matrix with raw read counts should be provided.
In case if normalized data are provided, the input file name should include "normalized", e.g.
pbmc3k_normalized.txt, therefore, no normalization
(such as SCTransform or log-normalization) will be applied.

The
first column of input file should correspond to gene annotations
(e.g. gene symbol, ENSEMBL Gene ID, etc) and should be called either
GENE_NAME or kept without any name.
Other columns are expected to correspond to cell/barcodes annotations.
The example data file can be found
here or downloaded on the "Upload" page.
The expected file extensions are either
*.xlsx, comma or tab-delimited
*.csv, or comma or tab-delimited
*.txt file (see example data).
For discrimination of mitochondria genes ScType allows the following prefixes:
-,
_, or
. (e.g.
mt-,
mt_,
mt.).
b. Output of the Cell Ranger pipeline from 10X Genomics.
Cell Ranger is a set of analysis pipelines that process Chromium single-cell RNA-seq output to align reads and generate gene-barcode matrices.
ScType will read in the output of the cellranger pipeline from 10X, and generate a unique molecular identified (UMI) count matrix from it, where the values of matrix represent number of molecules for each feature (i.e. gene; row) that are detected in each cell (column).
This matrix will be then used as if
Gene expression matrix input,
option a was provided.
The example data file can be found
here.
ScType expects *.zip archive comprising three output Cell Ranger files:
barcodes.tsv,
genes.tsv/features.tsv and
matrix.mtx.
Note: only Cell Ranger files should be archived, not the folder with files!

c. SingleCellExperiment R object
Note: This option is provided for users, who are familiar with a programming and want to upload their preprocessed data.
In case if SingleCellExperiment R object is provided, an *.RDS file with log-normalized read counts is expected by ScType. ScType will process the input file and look at
inputfile@assays$data$logcounts for the input matrix. No data normalization is applied when the file of this format is provided.
The example data file can be found
here or downloaded on the "Upload" page.
Uploading custom markers for cell type annotation
In order to upload the new markers for cell type annotation, please go to "Analysis results" page and follow the instruction on the figure below.

ScType will utilize the uploaded markers and make annotations based on them. To verify that the custom markers were uploaded correctly, click "upload custom markers" button again and check the table with currently used marker.
License
The ScType code is released under the GNU General Public License, version 3. Please follow this link for details.
Using ScType through R
An example can be found at GitHub.